Во месецот со најмалку дена во годината, во месецот кој пациентите со ретки болести го викаат „свој“, месецот кој е редок по своите ретки 28 или 29 дена ќе се обидеме поблиску да Ве запознаеме со ретките болести во Република Македонија.
Да ги дознаеме предизвиците и проблемите со кои се соочуваат. Како ја добиваат дијагнозата? Дали има некаков лек? Колку и дали воошто се пристапни и достапни лековите?…
Лоу синдром
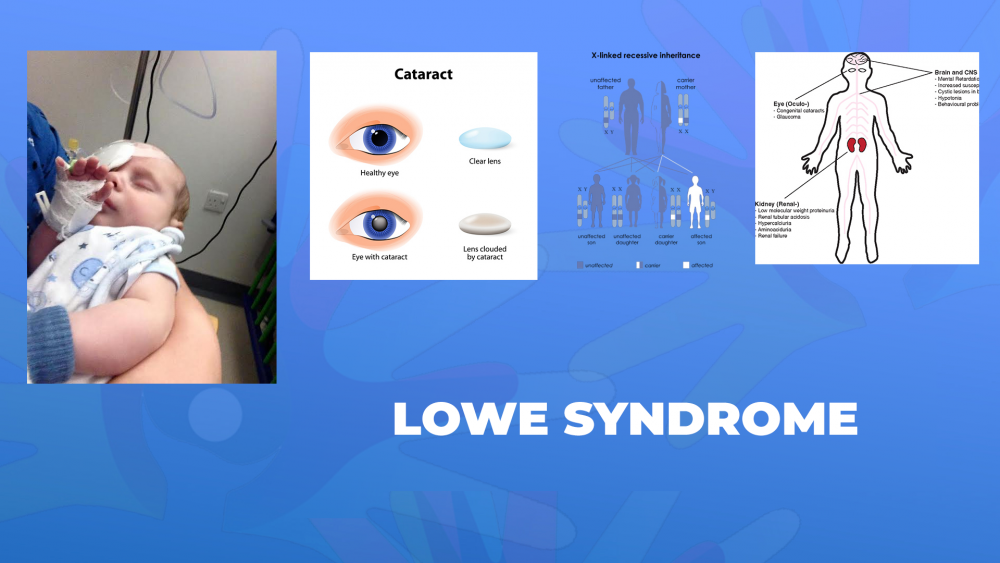
Лоу синдромот се карактеризира со проблеми со видот, вклучувајќи ја и катаракта која е присутна уште при раѓање, проблеми со бубрезите кои обично се развиваат во првата година од животот и абнормалности во мозокот кои се поврзани со интелектуална попреченост.
Начин на наследување
Лоу синдромот се наследува како Х-поврзана генетска состојба.
Клиничка слика
Новороденчињата со Лоу синдромот имаат катаракта која е присутна при раѓање (може да се открие со пренатален ултразвук кај сомнителните случаи). Корегираната визуелна острина ретко кога е подобра од 20/100.
Приближно кај половина од пациентите се јавува глауком кој може да доведе до слепило ако не се контролира. Новороденчињата имаат и слаб тонус на мускулите (хипотонија) при раѓање и задоцнет моторен развој. Се регистрира и развојна, интелектуална попреченост што може да варира од блага до сериозна форма. Парализите се јавуваат кај приближно половина од пациентите на возраст од шест години, а се јавуваат и промени во однесувањето. Кај овие пациенти се регистрира и Фанкониева проксимална тубуларна дисфункција. Оваа абнормалност резултира во губење на одредени материи (аминокиселини, бикарбонати и фосфати) во урината, кои обично се реапсорбираат пред екскреција во конечната урина. Други знаци кои се регистрираат при Лоу синдромот се низок раст, забни цисти и абнормално формирање на дентинот на забите, цисти на кожата и недостаток на витамин Д кои можат да доведат до меки коски, скелетни промени (рахитис), фрактури на коските, сколиоза и невоспалително дегенеративно заболување на зглобовите.
Причини
Синдромот Лоу е генетско нарушување поврзано со Х хромозомот, предизвикано од мутација во OCRL генот, што резултира во намалена активност на ензимот 5-фосфатаза. Околу една третина од заболените мажи имаат мутација. Кај останатите случаи, нарушувањето се наследува од страна на мајката, која е носител на генот. Секое девојче носител на оваа болест, на возраст над 10 години ќе покаже карактеристични промени во леќите на очите, различни од каква било друга метаболичка катаракта. Некои од нив ќе развијат значајна катаракта дури и во раните 30-ти години, доволна за да се побара оперативна интервенција. Истата може да биде пропуштена од оперативниот хирург. Како и да е, овие карактеристични промени во леќите на жената носител треба да го наведат офталмологот за да даде совети за идни бремености и добивање на новороденче од машки пол. Мажот има само еден Х-хромозом што го наследува од својата мајка. Ако новороденчето од машки пол наследи Х-хромозом што го содржи генот за овој синдром, тој ќе ја развие болеста.
Дијагноза
Лоу синдромот се дијагностицира кога намалената активност на ензимот 5-фосфатаза се покажува на култивирани клетки на кожата (фибробласти). Молекуларното генетско тестирање за мутации на OCRL генот е исто така достапно и точно се открива кај повеќе од 95% од заболените мажи. Околу 95% од девојчињата носители постари од 10 години имаат специфични и карактеристични абнормалности на леќата на окото што може да се дијагностицира од искусен офталмолог. Пренаталната дијагноза е достапна со биохемиско испитување (анализата на ензимите) или со молекуларно генетско тестирање, ако мутацијата на OCRL генот е утврдена кај заболен машки роднина или мајка носител.
Третман
Третманот на Лоу синдромот обично бара тим од медицински професионалци, вклучувајќи педијатриски офталмолог, нефролог, генетичар, нутриционист, ендокринолог, невролог, специјалист за развој на деца, општ хирург, ортопед и стоматолог.Слабиот тонус на мускулите (хипотонија) понекогаш може да резултира со проблеми во хранењето. Истиот може да биде причина за хранење со цевка и преземање на вообичаени мерки за гастроезофагеален рефлукс. Предвременото отстранување на катарактата се препорачува да се промовира оптимален развој на видот. Очилата за сонце и контактните леќи помагаат за подобрување на видот. Глаукомот што се јавува кај половина од мажите болни од овој синдром понекогаш може да се лекува медикаментозно, но обично е потребна операција. Фанкониевата проксимална тубуларна дисфункција се третира со орални додатоци од натриум и калиумбикарбонат и лицитрат. Дозите мора да се утврдат на индивидуална основа.
Оралниот фосфат и орален калцитриол се користат за лекување (или спречување) на рахитис. Густината на коските треба да се следи периодично. Нападите се контролираат со антиконвулзивни лекови. Проблемите во однесувањето се третираат со модификација на однесувањето и лекови. Се препорачуваат програми за рана интервенција кои вклучуваат физикална терапија, професионална терапија, говорна и јазична терапија, специјални служби за образование и услуги за лица со оштетен вид кои треба да започнат во раната детска возраст. Момчињата со Лоу синдромот треба редовно да се следат за проблемите со видот, функцијата на бубрезите, раст, напредокот, сколиоза и проблеми со зглобовите и забите. Бубрежната болест во крајната фаза успешно се лекува со дијализа и трансплантација на бубрег.
OhridNews