Конгенитална дискератоза (ДК) е ретка наследна форма на слабост на коскената срцевина, односно неможност на коскената срцевина да произведе доволно крвни клетки. Болеста зафаќа и други органи. Особено карактеристични се промените на формата и изгледот на ноктите (дистрофични нокти), променлива пребоеност на кожата (хиперпигментација) и бели наслаги на слузокожата во усната празнина (орална леукоплакија). Пациентите со конгенитална дискератоза имаат висок ризик за заболување и од други болести, вклучувајќи белодробна и црнодробна фиброза (замена на нормалното ткиво со сврзно ткиво), тумори, миелодиспластичен синдром, или леукамија. Болеста најчесто започнува во првата деценија од животот. Конгенитална дискератоза е ретка болест, со зачестеност од околу 4 нови случаи на 1 милион луѓе годишно.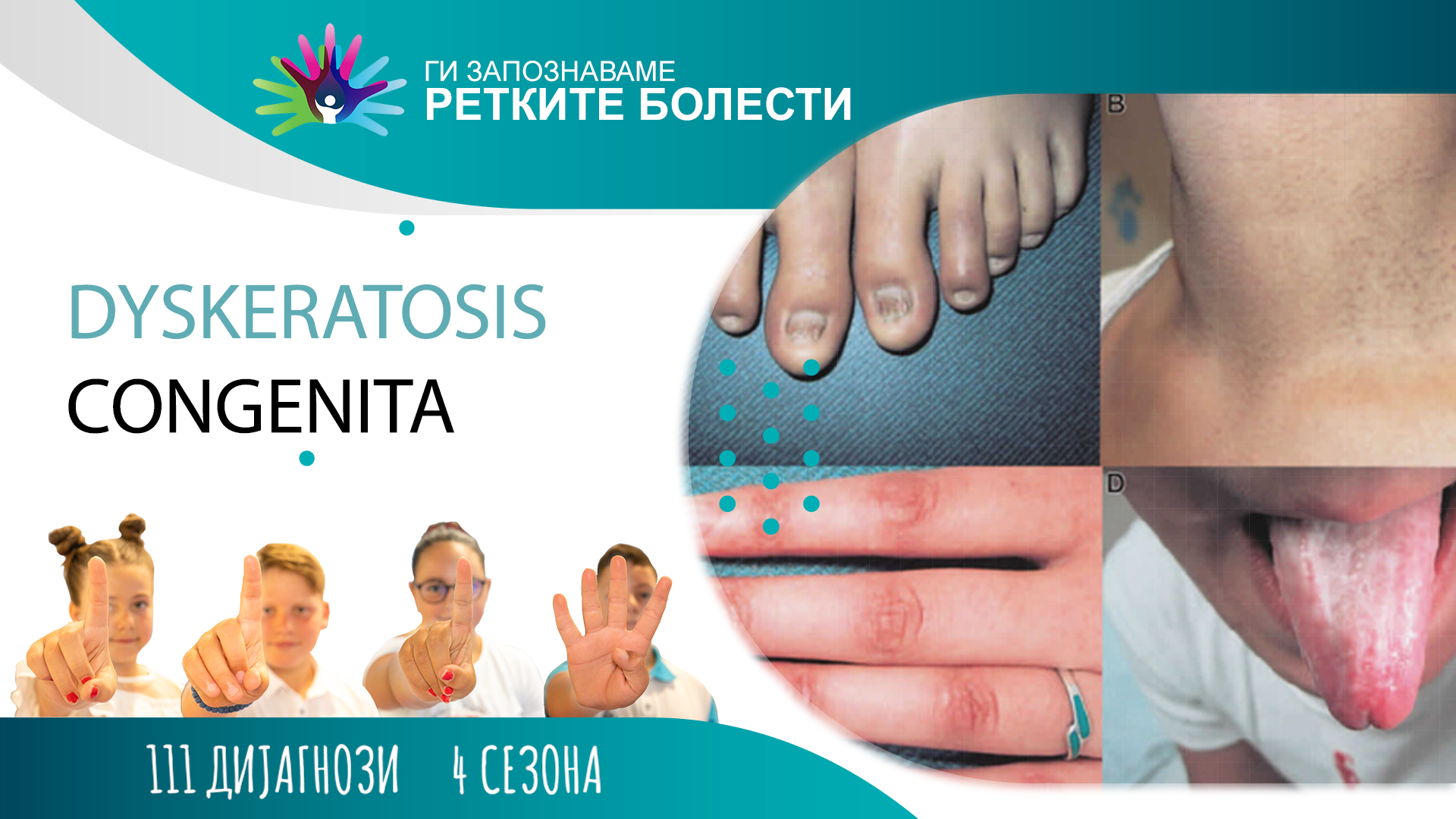
Симптоми:
Кај пациентите со Конгенитална дискератоза, клиничките симптоми и знаци се развиваат со различна брзина и на различна возраст, дури и во едно исто семејство. Кај класичната болест, промените на кожата и ноктите се јавуваат најрано и тоа во првата деценија, а 80% од пациентите развиваат слабост на коскената срцевина пред 30-годишна возраст. Околу 90% од пациентите ќе развијат слабост на коскената срцевина во некој одреден период од нивниот живот. Прв знак за слабост на коскената срцевина е намален број на тромбоцити (тромбоцитопенија), анемија, или пак и двете заедно. Потоа следи панцитопенија (намален број на сите крвни клетки) и апластична анемија (неможност за производство на сите крвни клетки). Кожните промени се карактеризираат со зони на потемна пребоеност (хиперпигментација) кои наликуваат на миле, и најчесто ги зафаќаат лицето, вратот, градниот кош и рацете. Степенот на пигментација се зголемува со возраста и може да ја зафати целата површина на кожата. Дистрофијата на ноктите вообичаено започнува со надолжно избраздување, расцепување или формирање на ноктен израсток и може да кулминира со целосен губиток на ноктите. Леукоплакијата вообичаено се јавува на јазикот, но може да се види и на другите слузници. Кај некои пациенти може да се појави прекумерно солзење (епифора), потешкотии со учењето и/или развојно заостанување, прекумерно потење на дланките и табаните, опаѓање и побелување на косата, кариес на забите, стеснување на хранопроводот, и др. Особено значајни компликации на Конгенитална дискератоза се појава на белодробна фиброза, црнодробна фиброза или гастро-интестинално крварење, кои се живото-загрозувачки состојби. Околу 10 % од пациентите, вообичаено во третата или четвртата деценија од животот, добиваат малигна болест, најчесто миелодиспластичен синдром, или акутна миелоична леукемија, канцер, т.е. рак на кожата или на гастро-интестиналниот систем.
Причини:
Конгенитална дискератоза е предизвикана од мутација во некој од неколкуте досега познати гени (DKC-1, TERC, TERT, TINF-2 и неколку други гени опишани само во поединечни случаи) кои доведуваат до промени во должината на теломерите. Теломерите се структури кои се наоѓаат на краевите од хромозомите и ги штитат од нивно слепување или расцепување. Прекумерното скратување на хромозомите доведува до забрзано стареење на клетката, нестабилност на генетскиот материјал и клеточна смрт. Ова се случува во сите клетки во организмот, но најзасегнати се оние со брза делба, меѓу кои се и клетките од коскената срцевина, кожата и слузокожите. Постојат Х-врзана форма (синдром на Зинсер-Кол-Енгман), автозомно доминантна форма и автозомно рецесивна форма на наследување, во зависност од засегнатиот ген. Кај некои пациенти, болеста се јавува спорадично што значи дека болеста не е пренесена од родителите. Само кај околу 70 % од пациентите се создава мутација во некој од познатите гени што укажува дека сè уште постојат неоткриени гени поврзани со Конгенитална дискератоза.
Дијагноза:
Дијагнозата за Конгенитална дискератоза се поставува според темелна клиничка евалуација, детална историја на пациентот и препознавање на карактеристичните промени, особено на кожата и во устата. Кај пациенти кои развиваат апластична анемија или белодробна фиброза, како прв знак на болеста, поставувањето на прецизна дијагноза е многу потешко. Од лабораториските анализи, еритроцитите се често макроцити (зголемени) и содржат повисоко ниво на фетален хемоглобин (форма на хемоглобин која се среќава во феталниот живот). Иницијалниот примерок од биопсија на коскена срцевина може да покаже нормална или зголемена клеточност, но со текот на времето настанува рамномерно опаѓање во сите крвни лози. Некои пациенти имаат имунолошки абнормалности, вклучувајќи намалена вредност на имуноглобулини и намален број на Б-лимфоцити и/или Т-лимфоцити. Цитогенетската анализа (анализа на генетскиот материјал во клетката) покажува многу кратки теломери во крвните клетки на пациенти со развиена слабост на коскената срцевина. Молекуларната генетска анализа за утврдување на мутации во засегнатите гени е клучна за поставување на прецизна дијагноза за Конгенитална дискератоза.
Терапија:
Андрогените можат да ја подобрат функцијата на коскената срцевина кај околу 70 % од пациентите со достигнување на нормален број на крвни клетки во текот на еден повеќегодишен период. Со прогресијата на болеста, одговорот на терапијата со андрогени се намалува. Терапијата со цитокини (мали молекули кои служат за сигнализација меѓу клетките), ГМ-КСФ или Г-КСФ, самостојно или во комбинација со еритропоетин (хормон кој стимулира производство на еритроцити), може да биде корисна, но само краткорочно. Трансплантацијата на коскена срцевина е единствената успешна опција кај пациенти со тешка апластична анемија, миелодиспластичен синдром или акутна миелоична леукемија. Долгорочното преживување на пациенти кај кои се направила трансплантација, дури и од сродни ХЛА-компатибилни донори, е ниско, значи само кај 50 % од пациентите. Високата стапка на смртност е последица на компликации поврзани со трансплантацијата или пак напредување на основната болест – појава на белодробна фиброза или гастро-интестинално крварење. Иако генетската основа на Конгенитална дискератоза е детектирана кај најголем дел од пациентите, сепак воведувањето на генска терапија е сè уште сон за многу од нив.
OhridNews